INTRODUCTION
L’artérite à cellules géantes (Giant cell arteritis des anglosaxons, maladie de Horton en France) est ainsi dénommée car on retrouve fréquemment dans la paroi des artères de grand calibre touchées un infiltrat inflammatoire à grands lymphocytes. C’est la plus fréquente des vascularites, touchant les sujets âgés de plus de 50 ans (incidence de 10 à 20/100.000/an dans cette population) et plus particulièrement les femmes. Elle donne classiquement une atteinte de l’extrémité céphalique (céphalées, claudication de la mâchoire, AIT, amaurose), des signes généraux ou rhumatologiques dans le cadre d’une pseudo polyarthrite rhizomélique et une atteinte des gros vaisseaux et en particulier de l’aorte thoracique (Large Vessels - GCA).
L’étude ARTEMIS est une étude française multicentrique destinée à faire une photographie des caractéristiques et de la prise en charge de cette pathologie en France.
METHODOLOGIE
Il s’agit d’une étude transversale multicentrique de recueil d’observations de patients de plus de 50 ans diagnostiqués avec une GCA sous traitement. Près de 3000 spécialistes en rhumatologie et médecine interne hospitaliers ont été sollicités pour inclure de manière consécutive jusqu'à 10 patients suivis pendant une période d’inclusion de 5 mois. Les données étudiées concernaient les méthodes ayant conduit au diagnostic, les comorbidités liées ou aggravées par la corticothérapie, les traitements administrés et l’activité de la maladie estimée indépendamment par le médecin et le patient sur une échelle visuelle analogique.
Le diagnostic de la maladie était considéré comme précoce, standard ou tardif selon que le temps écoulé entre les premiers symptômes et le diagnostic était inférieur à 1 mois, entre 1 et 3 mois ou de plus de 3 mois. L’analyse des données faisait également la part entre maladie incidente (phase aigüe, visite survenant < 6 semaines après le diagnostic) ou maladie prévalente au-delà.
RESULTATS
Soixante-neuf médecins spécialistes ont accepté de participer et d’inclure des patients au cours de l’année 2018. Il s’agissait de médecins internistes pour 84 % d’entre eux, rhumatologues pour 15 % et gériatres (1%).
Il est à noter que l’étude a été réalisée avant la reconnaissance de la spécialité de médecine vasculaire ce qui peut aussi expliquer l’absence de ces derniers (certains apparaissant sous l’intitulé internistes). Trois cent six patients ont été inclus.
Caractéristiques de la population : 72 % des patients avaient plus de 70 ans et la moyenne d’âge était de 74 ans, en grande majorité féminine (65%). Etant donné cet âge élevé au diagnostic, 83 % des patients étaient porteurs d’une comorbidité associée notamment l’HTA (46%), les dyslipidémies (12%) et le diabète (9.5%). La population étudiée dans l’étude était une population avec une maladie bien installée (moyenne de 13 mois depuis le diagnostic).
Mode de présentation initial
Quatre-vingt-neuf pour cent des patients avaient une atteinte de l’extrémité céphalique. Les céphalées étaient présentes dans 9 cas sur 10, une hypersensibilité du scalp et des anomalies cliniques des artères temporales dans un cas sur deux. Une atteinte oculaire à type de neuropathie ischémique antérieure aiguë était présente dans 10% des cas de même que la diplopie. Des symptômes de pseudopolyarthrite rhizomélique (PPR) étaient présents dans près de la moitié des cas (raideur matinale, douleur des ceintures). Les signes généraux étaient fréquents (fébricule et perte de poids chez un patient sur deux, altération de l’état général chez 9 patients sur 10). Au plan vasculaire, un anévrysme de l’aorte thoracique ou abdominale était présent chez 13 % des malades, une claudication des membres chez 18% des patients. Enfin et classiquement, un syndrome inflammatoire était très fréquent, VS > 50 mm à la première heure chez 77% des patients et CRP > 25 mg/l chez 84% des patients.
Le regroupement des symptômes est indiqué sur la figure ci-contre :
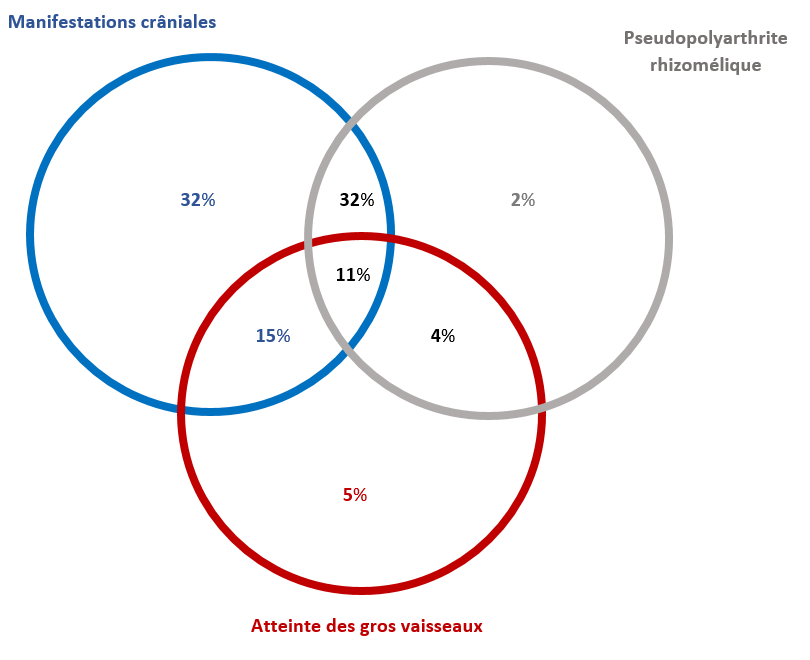
A noter que 5% des patients seulement avaient une forme isolée d’atteinte des gros vaisseaux.
Chez les patients avec une maladie prévalente et ayant connu au moins une rechute, la présentation initiale était plus fréquente avec des manifestations extra crâniales (sauf PPR).
Méthodes diagnostiques
Elles reposent sur l’association à l’examen clinique et à la biologie d’examens d’imagerie tels que l’écho-Doppler ou l’IRM des artères temporales, le PET scan ou d’anatomopathologie avec la biopsie des artères temporales. Cette dernière était la plus utilisée (85% des cas) et conduisait au diagnostic dans 67% des cas. L’écho-Doppler des artères temporales n’était utilisé que chez moins d’un tiers des patients (31%) et amenait au diagnostic chez 16% des patients. Le diagnostic était confirmé chez 54% des patients par la BAT, par l’écho-Doppler chez 16%, le PET scan chez 17%, l’angioscanner chez 8.8% et l’IRM chez 3% .Le PET scanner était plus utilisé que la BAT pour des patients avec une atteinte vasculaire et l’imagerie vasculaire était plus utilisée chez les patients avec un diagnostic tardif.
Rechutes et complications
La rechute reste fréquente dans la maladie puisque 46% des patients avec une maladie prévalente en avaient subi au moins une (57%) ou deux (26%). Le temps moyen de survenue de la rechute était de 10 mois. Seize pour cent des patients avaient au moins une complication après le diagnostic, de type ophtalmique (5%), psychiatrique (3%) et ou vasculaire (3%).
Traitement de la maladie de Horton
Tous les patients ont reçu une corticothérapie (CT) à un moment donné et 89% des patients étaient sous CT au moment de la visite de l’étude. La plupart des récidives ont été diagnostiquées chez des patients avec une dose moyenne de 10 mg de prednisone et étaient traitées avec une dose médiane de CT de 20 mg (10-30) dans 95% des cas. En addition, 29% des patients avaient un traitement en sus de la corticothérapie, immunosuppresseurs dans 19% des cas et/ou biothérapies dans 16% des cas. Les deux thérapeutiques adjuvantes les plus utilisées étaient le méthotrexate (19% des patients) et le tocilizumab (15%). Enfin 37 % des patients ont présenté au moins une comorbidité reliée ou aggravée par les corticoïdes à savoir diabète, HTA, ostéoporose et fractures.
DISCUSSION
Cette étude multicentrique française offre une photographie intéressante en 2018 d’un vaste échantillon de patients diagnostiqués avec une maladie de Horton. Au diagnostic, la grande majorité des patients (patientes surtout) est conforme à l’entité clinique de la maladie avec une symptomatologie oculo-cranio-faciale et une altération de l’état général. Le mode de diagnostic prédominant en reste la BAT (86%) même si l’écho-Doppler est réalisé chez 30% des patients. Il peut être espéré que le développement de spécialistes en médecine vasculaire puisse amener à augmenter ce chiffre et à épargner à certains patients un geste diagnostique invasif. Cette étude montre également que la présence de symptômes extra crâniaux (notamment vasculaires) est un facteur de risque de survenue de rechutes alors que celles-ci surviennent chez près de 50% des patients. La présence de formes vasculaires pures (aortites notamment) reste cependant faible, de l’ordre de 5%. Le haut pourcentage de rechutes sous corticothérapie de même que la prévention des complications de celle-ci explique que près de 30% des patients nécessitent un traitement épargneur de corticoïdes tel que le méthotrexate (19%) ou le tocilizumab (15%).
Référence
Protocole national de diagnostic et de soins (PNDS). Artérite à cellules géantes (Horton).2017. Révision (2020). Disponible en ligne : www.has-sante.fr/upload/docs/application/pdf/2017-08/pnds_-_arterite_a_c...